New method drives cellular HIV reservoirs to self-destruct
By Lauren Cahoon Roberts
The human immunodeficiency virus (HIV) is no longer a death sentence, yet a cure remains elusive. While current therapies can successfully manage active infection, the virus can survive in tissue reservoirs – including macrophage cells, which play an important role in the immune system.
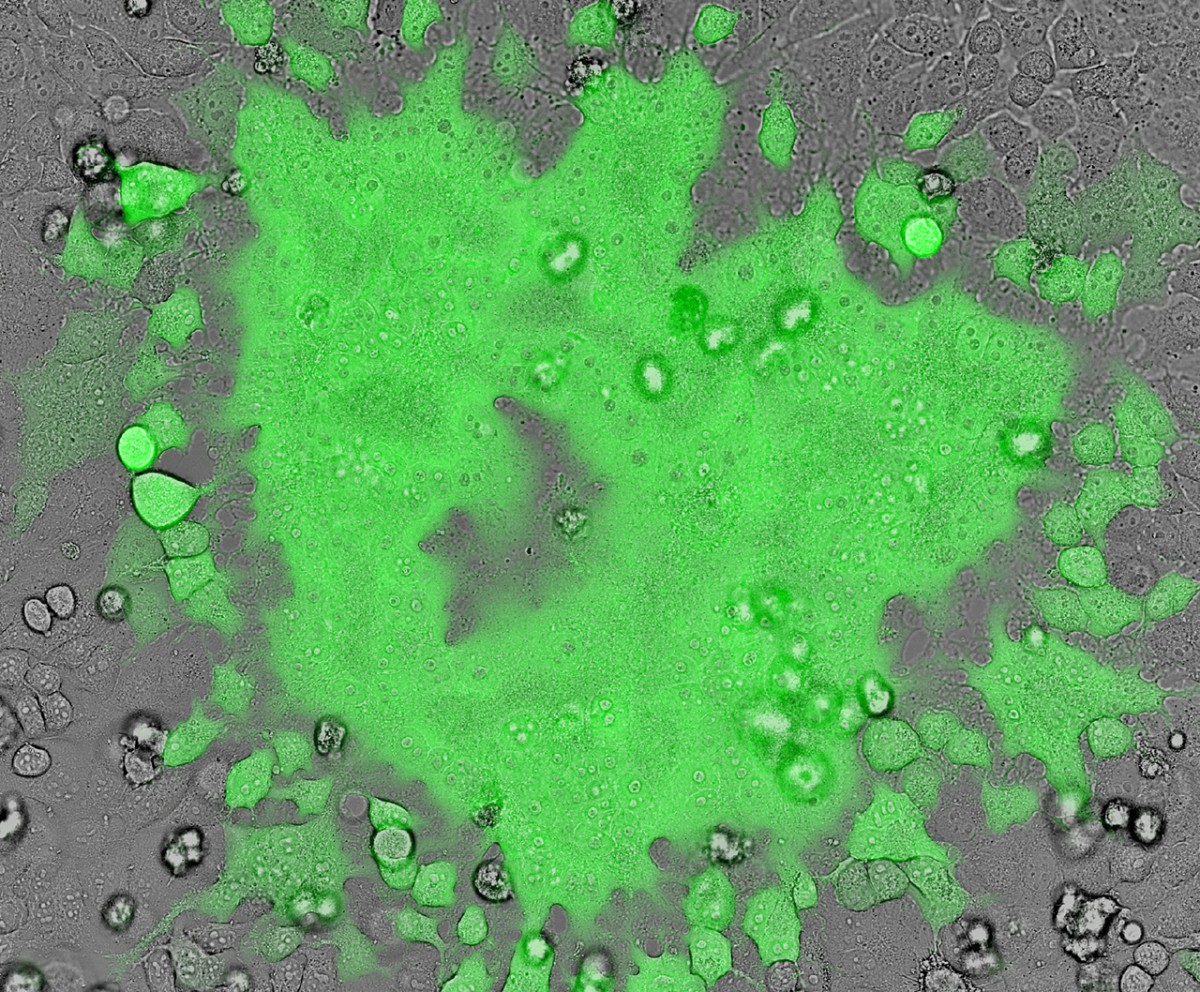
HIV-infected human macrophages co-incubated with a reporter cell line that only expresses GFP when it is infected with HIV-1. This shows that that the HIV-infected macrophages can transmit infection to susceptible bystander cells and is therefore a source of active infection.
David Russell, the William Kaplan Professor of Infection Biology in the College of Veterinary Medicine, and his research team in the Department of Microbiology and Immunology have pinpointed a novel angle of attack that could eradicate these viral reservoir cells – while leaving healthy cells untouched.
In their study, published March 27 in Proceedings of the National Academy of Sciences, Russell’s team, led by first author and postdoctoral fellow Saikat Boliar, describe how a genetic regulator called SAF helps HIV-infected macrophages avoid cell death. After blocking SAF in HIV-infected cells, the researchers found that these reservoir cells then self-destructed.
“We were all surprised by the specificity of the cell death,” Russell said. “Only infected cells die, while bystander cells, exposed to the same treatment at the same dose, showed no death at all.”
While macrophages – immune cells that consume foreign entities in the body – are helpful in fighting off certain microbes, they provide the perfect foxhole for HIV. Some researchers believe these infected macrophages are the reservoirs for persistent HIV infection.
“Current HIV drugs work really well on active infection, but it is the tissue reservoirs that are the problem,” Russell explained. “These sites of persistent virus are resistant to all current therapies.”
Russell, Boliar and their colleagues wanted to investigate what cellular mechanisms were at play that helped keep infected macrophages alive, and the researchers turned their attention to long noncoding RNAs (lncRNAs) – genetic coding elements that turn genes up or down, but do not translate directly into proteins themselves.
“We were interested in long-noncoding RNAs because they are known ‘master regulators’ of cell pathways, and had not really been looked at systematically in HIV infection,” Russell said.
The team screened a panel of 90 well-characterized lncRNAs in three distinct populations of human macrophages: healthy cells, HIV-infected cells and “bystander” cells – those that had been exposed to HIV but not infected.
The investigators found that SAF was more prevalent in the HIV-infected macrophages. Previous studies found SAF prevented self-destruction in cells. Russell and his team suspected SAF was protecting HIV-infected macrophages from dying.
To prove this theory, the team blocked SAF’s action using another noncoding RNA called small interfering RNA (siRNA), which effectively degrades targeted RNAs such as SAF. The researchers silenced SAF in the healthy, infected and bystander macrophage populations; the HIV-infected cells suddenly self-destructed, while the healthy and bystander cells remained unscathed.
“This showed us that when cells are infected with HIV, the virus alters the long noncoding RNAs’ expression in that cell,” Russell said. This would explain why bystander cells that are exposed to the HIV virions, but not actually infected by them, do not have the same response.
This discovery taps into a novel angle in curing HIV: selectively destroying persistently infected cells. The Russell team is eager to exploit it for potential therapies.
“We plan to do a drug screen for compounds that drive HIV-infected cells into programmed cell death,” Russell said. The team will start by looking for SAF inhibitors but also will look for other molecules that effectively eradicate reservoir cells through other mechanisms.
Co-authors include doctoral student David Gludish, from the Russell lab, and researchers from the Malawi-Liverpool-Wellcome Trust Clinical Research Program.
This work was supported by the National Institutes of Health.
Lauren Cahoon Roberts is assistant director of communications at the College of Veterinary Medicine.
Media Contact
Get Cornell news delivered right to your inbox.
Subscribe