Standard genome for heavily studied worm gets reboot
By Krishna Ramanujan
A new Cornell-led study finds that the genome for a widely researched worm, on which countless studies are based, was flawed. Now, a fresh genome sequence will set the record straight and improve the accuracy of future research.
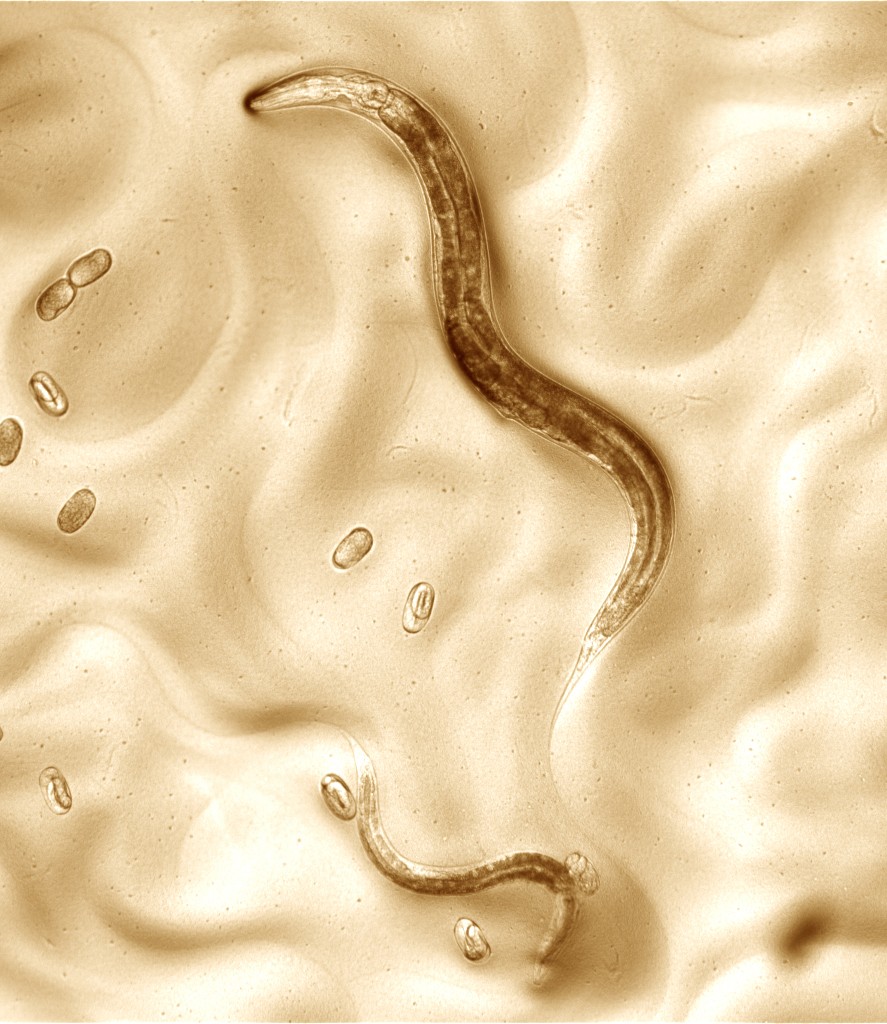
The nematode worm Caenorhabditis elegans. Three life stages of C. elegans are shown: an adult hermaphrodite, a smaller larva, and eleven newly laid eggs (with embryos developing inside them). These are all on a lawn of Escherichia coli bacteria (being eaten by the worms as food). The fully-grown adult is roughly 1 millimeter long.
When scientists study the genetics of an organism, they start with a standard genome sequenced from a single strain that serves as a baseline. It’s like a chess board in a chess game: every board is fundamentally the same.
One model organism that scientists use in research is a worm called Caenorhabditis elegans. The worm – the first multicellular eukaryote (animal, plant or fungus) to have its genome sequenced – is easy to grow and has simple biology with no bones, heart or circulatory system. At the same time, it shares many genes and molecular pathways with humans, making it a go-to model for studying gene function, drug treatments, aging and human diseases such as cancer and diabetes.
Genetic studies of C. elegans were based on a single strain, called N2, which researchers have ordered for decades from the C. elegans stock center at the University of Minnesota. Though people tried to uphold a common standard, individual labs grew N2 strains on their own, which led to morphing.
“Over the last decade, with more advanced genetic experiments using high levels of DNA sequencing, scientists were alarmed to discover that there is no longer a single laboratory strain that everyone was using,” said Erich Schwarz, assistant research professor in the Department of Molecular Biology and Genetics. “Over 40 years there have arisen many different N2 strains; we can’t rely on any one of them to do experiments.”
Schwarz is a senior author of a new study published May 23 in Genome Research that describes a single genetically clean strain, called VC2010, where each individual is truly identical. Schwarz and colleagues from the University of Tokyo, Stanford University, the University of British Columbia and the University of Minnesota used cutting-edge techniques to sequence VC2010’s genome and create a new standard.
“Before 2015, we could not have done the project we published just now,” Schwarz said. “The technology wasn’t there.”
Earlier iterations of genome sequencing technology could read up to 150 nucleotide letters (the basic structural unit of DNA) at a time, which were then stitched together to produce the full genome. The current tech provides more complete “long-read sequencing” that can at once read thousands of base pairs, even up to a million, from a single DNA molecule.
As part of the study, the researchers compared VC2010 to the original N2 genome. They expected a near-perfect match, but got a surprise. “Along with the 100 million nucleotides we expected to see, we discovered an extra 2 million nucleotides, an extra two percent of the genome,” that was hidden in the original, likely due to limitations of the old technology, Schwarz said.
In the past, people studying C. elegans genetics simply could not analyze the hidden sequences. “You are literally adding two percent,” Schwarz said. “If the mutation [you seek] happens to be within that hidden two percent, now you have a chance to spot it.”
Schwarz added that similar issues are likely occurring in the standard genomes of other organisms, including humans. “It shows us that having the true complete DNA of an animal is not as easy as we thought it was,” he said.
Other labs have begun using modern sequencing tools to reassess other genomes, which has implications for synthetic biology, where scientists are creating life – such as bacteria – from scratch. “Having a really good DNA sequence is an important baseline,” Schwarz said.
Lead authors include Jun Yoshimura and Kazuki Ichikawa in the lab of co-author Shinichi Morishita, professor of computational biology at the University of Tokyo, and Massa Shoura and Karen Artiles in the lab of co-author Andrew Fire, professor of pathology and genetics at Stanford University.
Media Contact
Get Cornell news delivered right to your inbox.
Subscribe